 Science
in Christian Perspective
Science
in Christian PerspectiveScience
in Christian Perspective
The Molecular Evolutionary Clock:A Critique
Gordon C. Mills
Department of Human Biological Chemistry and Genetics
University of Texas
Medical Branch
Galveston, TX 77555-0645
From: PSCF 46 (September 1994): 159-168
The molecular evolutionary clock hypothesis may be defined as the thesis that changes in amino acid sequence of a specific protein proceed at a constant rate in regard to time. Since it was proposed thirty years ago, this concept has been the primary molecular basis for evaluating organismal relationships (phylogenetic trees) and for estimating times of divergence of the various branches of those trees. We will consider recent developments in molecular biology and their relationship to the molecular clock concept. We will also discuss the lack of any theoretical basis for the molecular evolutionary clock and evaluate experimental data to show that there has been very little experimental verification of a constant rate of change. Finally, we will examine the relationship of the molecular clock hypothesis to some theological and philosophical beliefs.
The molecular evolutionary clock hypothesis may be defined most simply as the hypothesis that changes in amino acid sequence in a specific functional protein proceed at a constant rate in regard to time. The impetus for many of these protein sequence studies has been, as suggested by Jukes (1987, p. 87), that the theory provides a molecular means of measuring the course of evolution. The molecular evolutionary clock concept has been developed to the point that its precision has been considered by many to be of sufficient accuracy to be utilized as a means of calculating the time of divergence of homologous proteins. These divergence times and calculated branching points have been utilized in constructing PHYLOGENETIC TREES showing postulated ancestral relationships of known organisms (Fig. 1).
In the definition of
the molecular evolutionary clock hypothesis, it is important to note the words
"specific" and "functional" since the apparent rate of
change is different for different proteins, and the function of the protein has
been considered to be the primary constraint on the rate of change. When
phylogenetic trees are constructed using only MOLECULAR SEQUENCE DATA, the
greater the sequence distance, the greater the possible error in calculating
points of divergence (Romero-Herrera, et
al; 1979). As generally constructed, molecular phylogenetic trees often
utilize only the most parsimonious (i.e., economic) data. In this technique,
data are weighted to bring them into accord with morphologic and/or
paleontologic data, particularly for divergence points (Romero-Herrara, et al;
1979). Although initially the molecular clock concept was based on sequences in
functional proteins, and in genes for those proteins, more recently it has been
extended to include segments of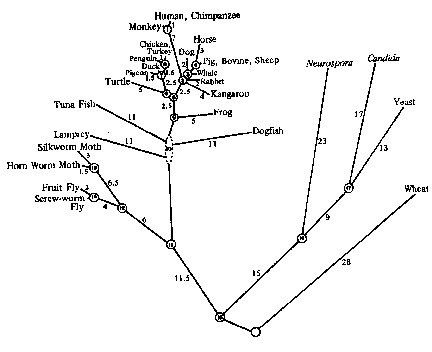 DNA (e.g., PSEUDOGENES, INTRONS, etc.) that are considered by some to be
nonfunctional.
DNA (e.g., PSEUDOGENES, INTRONS, etc.) that are considered by some to be
nonfunctional.
Historically, the concept of a constant change of rate with time was proposed by Ingram (1961) and Zuckerkandl and Pauling (1962) using amino acid sequences in proteins. Zuckerkandl (1987) has reviewed some historical aspects and conceptual perspectives of the molecular evolutionary clock. As techniques for sequencing NUCLEOTIDE units in DNA became available, the concept was extended to include DNA sequences. This would be a very logical extension, since the genetic information for protein sequences resides in CODING SEQUENCES of DNA. However, DNA is made up of more than coding sequences, and hence DNA sequence studies give a much broader picture, but also one that is much more complex. It should be emphasized that both protein sequence comparisons and DNA sequence comparisons deal with only a tiny fraction of the GENOMES of organisms.
More recently, DNA hybridization techniques have been developed that compare much larger portions of the genomes of organisms. However, these techniques suffer from being much less precise than amino acid or nucleotide sequence studies. Hybridization studies are based on the long known principal that double-stranded DNA will separate into single strands with an increase in temperature as a consequence of breakage of hydrogen bonds. On slow cooling, the two strands will come back together in the form of a double helix. If one mixes single-stranded DNA from two different organisms, the extent of formation of double helices depends on the extent of similarities in DNA molecules of the two organisms.
There are many other types of studies that provide comparisons between organisms at the molecular level (e.g., ELECTROPHORETIC comparisons of proteins, studies of metabolic pathways, immunologic studies, etc.). These have been of great value for taxonomic comparisons of organisms, but most of these have not been utilized as extensively for the molecular evolutionary clock hypothesis, so they will not be considered further here.
The Molecular Evolutionary Clock: Is There a Theoretical Basis?
There has been no adequate proposal of a theoretical basis of the molecular evolutionary clock hypothesis. I will consider this aspect of the hypothesis from the standpoint of (1) rates of occurrence of mutations, (2) establishment of mutations in the genome, (3) change with chronological time vs. generation time, and (4) change as a consequence of single mutational events.
(1) It should be noted that there is no sound theoretical basis for a constant rate of incorporation of mutations in genes for a particular protein. There are multiple causes of mutations, ranging from exposure to ultraviolet light and to radiation from radioactive isotopes, to exposure to mutagenic chemicals from air, water or diet. None of these would be expected to be constant with respect to time in all geographical areas, or under all environmental conditions. During replication, DNA is copied with an amazing degree of fidelity, but occasionally copying mistakes will be made, leading to point mutations, or possibly to deletions or insertions. It is possible that this type of mistake could have a relatively uniform rate even among different species. However, in vitro, the rate of copying mistakes varies quite markedly and depends on pH, ionic strength, ion concentrations, etc of the environment. It seems likely that both external and internal environmental conditions might affect the rates of copying errors. Other mutational events may occur at the time of cell division as a result of gene conversions. However, there is no reason to believe the rate of these events would be constant as they relate to chronological time.
There is also another important factor that affects mutation rates. Cells have the capacity to repair most mistakes in DNA, whether mistakes are made prior to or during the replication process. Neel (1982) notes that at least eight different types of repair systems have been demonstrated. Some of these utilize excision of damaged bases followed by replacement with the correct base and subsequent ligation of the broken DNA strand. In general, these repair systems are rather sophisticated, utilizing several enzymes, coenzymes and energy resources of the cell. Recent studies have indicated that these repair mechanisms may have specificity for certain genes or for certain regions of chromosomes, and may respond to specific signal sequences. DNA repair is of great significance in reducing the incidence of malignant tumors in humans (as well as in other organisms), but for this paper we are more interested in DNA repair in GERMINAL ORGANS (ovaries and testes) that would minimize the number of mutations that are transmitted from parents to offspring. There is no reason to believe that the overall effect of DNA repair would permit a uniformly constant rate of mutations.
(2) Once a mutation has occurred, its establishment in the genome for a particular species would be a very rare event. Factors such as geographic or reproductive isolation of a very small population with the mutant gene would play a major role in the rate of establishment of the mutant gene in the genome. There is clearly no theoretical reason why these processes should yield a constant rate. It is widely accepted that a gene for each protein has its own rate of mutation with regard to time. The differences in mutation rates are generally attributed to the presence or lack of functional restraints in the protein molecule, a concept which we will discuss separately. Classification of mutation rates for different types of gene segments is arbitrary. Generally, coding sequences of genes are expected to have lower mutation rates than introns or noncoding sequences because the latter are considered to be less functional.
(3) As has been pointed out by many others, if the mutation rate is constant, it should be related to generational time, rather than chronological time (Williams, 1974). This is a consequence of finding that most mutations are incorporated into the genome at the time of cell division. There is obviously a tremendous variation in generational times for different organisms. Since we are dealing with passage of mutations from one generation to the next, the cell divisions would be those of germinal cells (ova or sperm or precursors of ova or sperm).
(4) Inherent in the concept of a molecular clock is the idea of gradual change, i.e., one mutational event at a time. If the change is indeed gradual, we should expect to find a great many more intermediate (i.e., very closely related) informational molecules in a single species. Although there are a few closely related molecules (e.g., the gammaG and gammaA GLOBINS, which differ in only one amino acid), in many cases the informational molecules for different ISOZYMES within a particular species are quite divergent. This is illustrated by the two different cytochrome c isozymes in the mouse, or the two different cytochrome c isozymes in the fruit fly (Mills, 1992), or by the marked sequence differences in alpha, beta and gamma globins. It is clear that the evidence for very gradual change (e.g., establishment of single mutational events) is not present in genomes of organisms. Does this suggest that very closely related molecules tend to be eliminated from the genome or be corrected (repair mechanisms, gene crossovers, etc.)? Neel (1982) has argued strongly that corrective mechanisms play a major role in preventing change in the cellular genome.
Functional Constraints and a Molecular Clock
One facet of the molecular evolutionary clock theory that has been recognized by all proponents of the theory has been that the rate of sequence change was different for every protein. For example, according to the theory, the time required for a 1% change in sequence of proteins is 20 x 106 cytochrome c, 5.8 x 106 years for hemoglobin, and 1.1 x 106 years for FIBRINOPEPTIDES (Dickerson, 1971). Thus, amino acids in fibrinopeptides change twenty times as rapidly as do those of cytochrome c. Others have noted that HISTONES have the slowest rate of change. Histone H4 has about one-thirtienth the rate of change of cytochrome c (Behe, 1990). These differences in rates of change have traditionally been attributed to differences in functional constraints built into the three-dimensional structures of protein molecules. Nevertheless, until recently, the concept of functional constraints had not been subjected to any quantitative test.
In recent years, techniques have become available to modify genes at specific sites and determine whether the modified genes (and the corresponding modified proteins) would function in cells. Behe (1990) notes that Grunstein's studies show that histone H4 may still function when as many as ten amino acids are deleted. This clearly shows that the concept of functional constraints cannot be applied to histones. Whether the concept of functional constraints still has merit for explaining the differences of evolutionary rates of cytochrome c and hemoglobin is an open question.
Despite our knowledge of the three-dimensional structures of cytochrome c and hemoglobin, no one has been able to show quantitatively that the cytochrome c molecule is more functionally constrained than the hemoglobin molecule. Consequently, the concept of functional constraints as an explanation for differences in rates of change in protein molecules still lacks experimental support.
Modern Molecular Biology Developments
Let us examine briefly some developments in modern molecular biology that are important in understanding literature related to the molecular evolutionary clock hypothesis. With a genetic code of three PURINE AND PYRIMIDINE BASES per CODON and four different bases in DNA, there are 64 different possible combinations of three bases (i.e., 64 different three letter codons). Since there are only 20 amino acids to be coded, plus INITIATION and TERMINATION CODONS, there is more than one codon per amino acid (Fig. 2). Consequently, the number of codons per amino acid range from one each for methionine and tryptophan to six each for leucine and arginine, with two to four codons for each of the other amino acids.
With a few exceptions, the different codons for an amino acid differ in the third position of the codon. Consequently, we can experimentally find differences in third positions of codons in a coding sequence of a gene for a specific protein without noting any change in amino acids. These are referred to as synonymous changes in the gene; when the codon change would cause an amino acid change, it is referred to as a nonsynonymous change.
Simply from examining the genetic code (Figure 2), it is evident that purine to purine, or pyrimidine to pyrimidine changes (TRANSITIONS), would be more apt to be synonymous than purine to pyrimidine, or pyrimidine to purine changes (TRANSVERSIONS). For example, a change in a phenylalanine codon from TTT to TTC would be a transition and a SYNONYMOUS CHANGE, while a change from TTT to TTG would be a transversion and a NONSYNONYMOUS CHANGE. (TTG is a codon for the amino acid leucine.) Rats and mice, two rodents, have identical amino acid sequences in cytochrome c, but the nucleotide sequences in coding regions of the genes for cytochrome c differ in nine positions. These differences are all in third positions of codons and are all synonymous in nature.
Various equations have been devised to correct for multiple hits, but all are only approximations. It should be emphasized that the larger the correction for multiple hits, the larger the possible error.
In comparing sequences, it is also important to note that for any given point in a gene, there may have been more than one change, referred to as multiple hits (e.g., A - >G - > C, where all that we may note experimentally is the A -> C change). Multiple hits may also result in back mutations. A back mutation being one where an initial change is reversed by a subsequent mutation (e.g., A - > G - > A). Since the overall effect of this would be no change, a back mutation would not be detectable. Theoretically, one fourth of multiple hits would result in back mutations.
Various equations have been devised to correct for multiple hits, but all are only approximations. It should be emphasized that the larger the correction for multiple hits, the larger the possible error. When no corrections in data are made for multiple hits, the convention is to use the term "differences" in comparisons of genes or protein sequences; when multiple hit corrections are made, the term "substitutions" is utilized. Most recent papers conform to these usages, although earlier papers may not.
It is very important to note various types of changes that may occur in genetic material. First, point mutations involve a change in a single nucleotide base of DNA (or possibly several bases). Causes of point mutations were discussed earlier. Point mutations would usually cause either no change in amino acid sequence in the expressed protein or a single amino acid change. Occasionally, a point mutation might introduce a termination codon, which would terminate any synthesized polypeptide at that point. If the mutation involved nucleotide insertions or deletions, unless the event involved three (or a multiple of three) nucleotides, the coding sequence from that point on would change (this is called a frame-shift), and any expressed protein would have no similarity from that point on to the protein expressed by the gene prior to the insertion or deletion. If the insertion or deletion involved three (or a multiple of three) nucleotides, there would be an insertion or deletion of an amino acid(s) in the expressed protein.
Other types of intraspecies changes are often grouped together as gene conversions. They are of various types, but all would involve transfer within an individual of much larger portions of genetic material. For example, gene crossovers might involve a transfer of a DNA segment containing as little as only a portion of a gene (but many nucleotide units) up to many genes. The transfer might be from one chromosome to another or within a single chromosome. It should be immediately evident that transfers of large segments of genes would markedly complicate any interpretation of molecular evolutionary clocks involving those genes. In most comparisons of nucleotide sequences in genes, one can rule out the possibility of transfers of large gene segments. If the transfer involved a small gene segment (e.g., eight to fifteen nucleotides), it might be more difficult to detect.
Syvanen (1987) discusses evidence for the interspecies transfer of genetic information. In most cases, the transfer involves RNA viruses known as RETROVIRUSES. It has long been known that portions of viral genomes may appear in mammalian genomes. More recently, however, it has been demonstrated that retroviruses may also carry an occasional host gene along with the viral gene when they are transferred. Thus there is the possibility of transfer of a gene or a gene segment from one organism to another of the same species or even to a different species. Because of host-viral specificities, this type of interspecies gene transfer would occur most often in closely related species. However, there are some retroviruses that may infect a wide variety of mammalian species, so there is a possibiity of gene transfer between quite diverse species (e.g., mouse to man).
Some molecular evolutionary clock studies have utilized an inactive type of gene known as pseudogenes as a means of estimating diversion dates and in establishing phylogenetic trees. (For a discussion of pseudogenes, see Mills; 1992). However, pseudogenes are particularly prone to interspecies transfer of the type described above. Hence, molecular evolutionary clocks based on pseudogene studies are even less likely to be reliable. The possibility of interspecies gene transfer also opens its use as an explanation for discarding results that are not in accord with the molecular evolutionary clock theory.
In closing this part of the discussion, we can note that there are many experimental studies showing that point mutations occur (e.g., see my review of the mutations occurring in human hemoglobins (Mills; 1975)); gene conversions also are well established experimentally. Interspecies gene transfer studies are relatively new and the extent of these is still an open question. That they do occur, however, appears to be beyond doubt. The question is not whether mutational events of the types discussed do occur; they clearly do. The question is whether, or to what extent, mutational events can account for ancestral relationships, and what is their value in establishing phylogenetic relationships. The question must be asked: Does an amino acid difference or a nucleotide difference in comparison of proteins or in comparison of genes provide proof of a point mutational event, or is it only an indication that there may have been a past point mutational event? This distinction is very important, and conclusions reached in discussing either ancestral or phylogenetic relationships are dependent in considerable degree on our answers to those questions.
Experimental Data: Does It Support a Constant Rate of Change?
Although the proposal
of the molecular evolutionary clock was made initially by Zuckerkandl
and Pauling (1962) and by Ingram (1961), Dickerson
(1971) provided the first thorough examination of protein sequence data in
relation to evolutionary time. Plots of his data appear to show a linear
relationship between amino acid changes and times of divergence for cytochrome
c, the globin chains, and fibrinopeptides. A replot of Dickerson's cytochrome c
data on a slightly expanded scale (Figure 3) indicates that there are some
points that differ markedly from linearity. For example, cytochrome c amino acid
changes per 100 residues comparing primates and other mammals is given as 9.7,
with a divergence date of 90 million years, while the corresponding amino acid
change is 9.5 when mammals and birds are compared. However, the listed
divergence date in the latter instance is 300 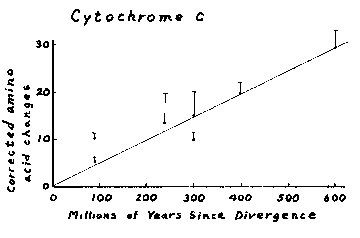 million
years. It should be immediately evident that this would represent over a
three-fold variation in the rate of change.
million
years. It should be immediately evident that this would represent over a
three-fold variation in the rate of change.
Romero-Herrera, et al (1979) noted considerable variations in rates of changes in amino acid sequence for particular proteins. For example, myoglobin amino acid sequences of the pig and the bat differ by 9, whereas the corresponding sequences of the more closely related pig and ox differ by 28. There is no reasonable explanation for this unusual finding, which clearly contradicts the concept of a molecular evolutionary clock.
Baba, Darga, Goodman and Czelusniak (1981) tabulated the changes in cytochrome c using all known sequences and the best available paleontological data. They found that nucleotide replacements per 100 codons per 100 million years for cytochrome c has varied from 1.4 to 17.3.
Jukes (1987) has noted the nonconstancy of the molecular clock for cytochrome c in different species. He and Holmquist (1972) noted that the rate of replacement of amino acids was nearly twice as fast in the rattlesnake as in another reptile, the snapping turtle. Beintema and Campagne (1987) utilized amino acid sequences of insulin in 10 species of rodents to prepare phylogenetic trees. The evolutionary rate of change was comparable to that of other species in the rat, mouse and hamster. However, they found moderately increased evolutionary rates in the porcupine and chinchilla, and markedly increased evolutionary rates in the guinea pig, cuis, copyu and casiragua. The differing rates made it nearly impossible for them to produce reasonable phylogenetic trees for these rodents, and the authors present three very different rodent phylogenetic trees.
Britten noted that rates of change in DNA among these various taxonomic groups differ by a factor of five.
Schwabe (1986) has examined amino acid sequences in the hormone relaxin in six species. Relaxin is a hormone that widens the birth canal during parturition. It is a polypeptide with some sequence similarity to insulin. Yet amino acid sequences of relaxin do not fit any possible phylogenetic tree. For example, pig relaxin is more similar to that of sharks and snake than it is to relaxin of the rat.
Shaw, Marks, Shen and Shen (1989) have studied nucleotide sequences of the alpha-globin gene in a number of primates. They noted a burst of evolution in nonsynonymous sites of the alpha-globin gene of baboons subsequent to their separation from the rhesus monkey. There was no corresponding increase in the rate of change for synonymous sites of the alpha-globin gene.
Catzeflis, Sheldon, Ahlquist and Sibley (1987) utilized DNA:DNA hybridization studies to estimate relationships among eight species of arvicoline rodents (mostly voles) and six species of muroid rodents (mostly rats and mice). They estimated the rate of divergence in these rodents as about 10 times the rate of divergence in hominoid primates.
Britten (1986) summarized DNA sequence data from a wide variety of taxonomic groups. He considered silent subsitutions (synonymous substitutions) in coding sequences of DNA. He also compared the data with that obtained from DNA:DNA hybridization studies. He noted that rates of change in DNA among various taxonomic groups differ by a factor of five. The slowest rates were noted for higher primates and some bird lineages, while faster rates were seen in rodents, sea urchins and Drosophila. Caccone and Powell (1990) utilized DNA:DNA hybridization studies to estimate rates of change in DNA of Drosophila (fruit flies). They concluded that the rate of change was five to ten times faster in fruit flies than for most vertebrates.
Brunk, Kahn and Sadler (1990) studied differences in amino acid sequences in H3II and H4II histones of nineteen species of Tetrahymena (a ciliate protozoan). In most other species, histones have an extremely low rate of change. However, these authors found much greater rates of change in the histones of ciliate protozoans. In regard to ciliates, Syvanen notes: "The ciliates differ so much that the molecular clock calculations place divergence of Tetrahymena, for example, back to more than 3 billion years ago; a clearly absurd result" (1986, p. 64). Hickey, Benkel, Boer, Genest, Abukashawa and Ben-David (1987), utilizing DNA sequences of alpha amylases, found substitution rates varying by as much as tenfold. In a comparison of viral oncogenes, Gojobori and Yokoyama (1987) noted that the rate of substitution in viral oncogenes was one million times that of their human counterpart.
Scherer: "It (the protein molecular clock) can neither be used as a tool for dating phylogenetic splits nor as reliable supportive evidence for any particular phylogenetic hypothesis...It is concluded that the protein molecular clock hypothesis should be rejected."
Scherer (1990) has recently provided a critical evaluation of the protein molecular clock hypothesis. After reviewing data for eight different types of protein molecules, he concludes: (p. 102, 103) "It (the protein molecular clock) can neither be used as a tool for dating phylogenetic splits nor as reliable supportive evidence for any particular phylogenetic hypothesis...It is concluded that the protein molecular clock hypothesis should be rejected." Various authors have proposed means of testing whether a particular protein clock is a "good" clock or a "poor" clock. Scherer (1990) has also reviewed data from these tests and concludes that they are not valid for testing usefulness of a particular protein clock.
The references cited above should clearly demonstrate the lack of constancy in rates of change in genomes of organisms. This lack of constancy puts investigators in the peculiar position of using data if it agrees with the molecular clock hypothesis, or discarding it or explaining it away if it does not agree with the hypothesis. Surely this cannot be legitimate science!
We must question whether sequence studies have a legitimate role in future studies of genetic relationships. Some authors have insisted that they have a role in studying relationships of closely related species. Yet, as noted above, some of the greatest variations in evolutionary rates have been shown in closely related species (e.g., rodents). At best, it would seem that sequence studies should be interpreted cautiously and only in relation to morphologic comparisons of the same organisms. In my view, nucleotide or amino acid sequence studies have little or no value in the estimation of divergence dates of ancestral organisms, and only limited value in identifying ancestral relationships. Their use in constructing phylogenetic trees for the entire animal kingdom is so subject to error that the generated trees have little value. This is because the calculation of branching points in molecular phylogenetic trees assumes a constant rate of divergence. If the rates of divergence are not constant, then calculated branching points are not likely to be correct!
Theological Implications of the Molecular Evolutionary Clock Theory
For many advocates of scientific naturalism, molecular evolutionary clock theory is a cornerstone of their belief in ancestral relationships, i.e., that every organism on earth today is a descendent of one archtypal organism. In other words, ancestral relationships of all organisms may be represented in a branching phylogenetic tree (Figure 1, for example). Moreover, all branches in the tree are a consequence of chance events, with natural selection being the driving force for diversification.
This role of the molecular evolutionary clock theory is illustrated by Thomas Jukes (1990) in his rebuttal to Phillip Johnson's (1990) arguments on the establishment of naturalism. To quote Jukes:
As more hemoglobin molecules became sequenced, the steady increase in divergence, calculated from reference points in the fossil record, showed evidence of a so-called molecular evolutionary clock. The same evidence was found in another family of proteins, the cytochromes c, and this made it possible to conclude that the common ancestor of yeast, plants, and vertebrates lived about 1.2 billion years ago. (Jukes, 1990, p. 17)
Surely this statement by Jukes indicates a commitment to scientific naturalism, when he proceeds to extrapolate from limited data to the grand theme of that philosophy. This interpretation of the above statement by Jukes is in complete accord with his earlier statement (Jukes, 1987, p. 87) that the molecular evolutionary clock provides a molecular means of measuring the course of evolution.
Despite claims of the more extreme evolutionists that all organisms (and hence all genetic material in these organisms) are products of natural causes, there has been no scientific explanation for the formation of new genetic information.
Quotations from Holmes Rolston (1992) provide an indication of the changing views of some scientists: "In stark contrast to divine design, natural selection is blind. Random genetic variations that are accidentally useful are selected; the most worthless are discarded." Rolston then continues with suggestions of purpose and possibly even design: "Contemporary geneticists are insisting that, though not deliberate, the process is cognitive. A vast array of sophisticated enzymes cuts, splices, rearranges, mutates, reiterates, edits, corrects, translocates, inverts and truncates particular gene sequences."
Despite claims of the more extreme evolutionists that all organisms (and hence all genetic material in these organisms) are products of natural causes, there has been no scientific explanation for the formation of new genetic information. In fact, as has been shown mathematically by Yockey (1977), even the chance formation of a single enzyme protein such as cytochrome c is beyond the realm of possibility (probability is 2 x 10-65). Even the simplest organisms have thousands of different proteins. Eukaryotic organisms (organisms with nucleated cells) are far more complex than prokaryotic cells (e.g., bacteria, etc.); vertebrates are more complex than invertebrates, etc. This increase in complexity comes as a consequence of new genetic information, but from where does this new genetic information come? Even if the molecular evolutionary clock theory were valid, it omits entirely the question of the origin of new genetic information, since any concept of design is considered to be outside the realm of science.
As a Christian and as a scientist, my belief is that I should leave open the possibility that God in his sovereignty may have chosen to provide genetic information as needed, thus guiding the diversification of species that one sees today and which is evident throughout the fossil record. In the molecular evolutionary clock theory, I see an effort by some scientists to force data into a mold where it doesn't really fit. Melnick (1990) has summarized the proceedings of a recent conference that considered the molecular evolutionary clock hypothesis. His review provides an indication of the diversity of opinion on the subject, and also of the desire by many to hold on to some remnant of the hypothesis, even though nearly all agree that there is no constant rate of change. My faith in the integrity of science has been partially restored when I see that papers are now being accepted in the scientific literature that demonstrate the failures of the molecular evolutionary clock hypothesis. More often than not, however, the authors consider these as exceptions to the rule rather than as an invalidation of the original hypothesis.
©1994
References
Baba, M.L., Darga, L.L., Goodman, M. and Czelusniak, J. (1981) "Evolution of Cytochrome C Investigated by the Maximum Parsimony Method." J. Mol. Evol. 17: 197-213.
Bientema, J.J. and Campagne, R.N. (1987) "Molecular Evolution of Rodent Insulins." Mol. Biol. Evol. 4: 10-18.
Behe, M.J. (1990) "Histone Deletion Mutants Challenge the Molecular Clock Hypothesis." Trends Biochem. Sci. 15: 374-376.
Britten, R.J. (1986) "Rates of DNA Sequence Evolution Differ between Taxonomic Groups." Science 231: 1393-1398.
Brunk, C.F., Kahn, R.W. and Sadler, L.A. (1990) "Phylogenetic Relationships among Tetrahymena Species Determined Using the Polymerase Chain Reaction." J. Mol. Evol. 30: 290-297.
Caccone, A. and Powell, J.R. (1990) "Extreme Rates and Heterogeneity in Insect DNA Evolution." J. Mol. Evol. 30 273-280.
Catzeflis, F.M., Sheldon, F.H., Ahlquist, J.E. and Sibley, C.G. (1987) "DNA:DNA Hybridization Evidence of The Rapid Rate of Muroid Rodent DNA Evolution." Mol. Biol. Evol. 4: 242-253.
Dayhoff, M.O. (1969) Atlas of Protein Sequence and Structure. Washington, D.C., National Biomedical Research Foundation, Vol. 4.
Dickerson, R.E. (1971) "The Structure of Cytochrome C and the Rates of Molecular Evolution." J. Mol. Evol. 1: 26-45.
Gojobori, T. and Yokoyama, S. (1987) "Molecular Evolutionary Rates of Oncogenes." J. Mol. Evol. 26: 148-156.
Hickey, D.A., Benkel, B.F., Boer, P.H., Genest, Y., Abukashawa, S. and Ben-David, G. (1987) "Enzyme-Coding Genes as Molecular Clocks: The Molecular Evolution of Animal Alpha-Amylases." J. Mol. Evol. 26: 252-256.
Ingram, V.M. (1961) "Gene Regulation and the Hemoglobins." Nature 189; 704-709.
Johnson, P.E. (1990) "Evolution as Dogma: The Establishment of Naturalism" First Things, No. 6 (October): 15-22.
Jukes, T.H. (1987) "Transitions, Transversions and the Molecular Evolutionary Clock." J. Mol. Evol. 26: 87-98.
Jukes, T.H. (1990) "A Response to Phillip Johnson." First Things, No. 6 (October): 26-27.
Jukes, T.H. and Holmquist, R. (1972) "Evolutionary Clock: Nonconstancy of Rate in Different Species." Science 177: 530-532.
Melnick, D.J. (1990) "Molecules, Evoluton and Time." Trends Ecol. Evol. 5: 172-173.
Mills, G.C. (1975) "Hemoglobin Structure and the Biogenesis of Proteins. Relation of Structure to Function for Mutant Hemoglobins." J. Amer. Sci. Affil. 27: 33-38.
Mills, G.C. (1992) "Structure of Cytochrome C and C-like Genes: Signigicance for the Modification and Origin of Genes." Perspec. Sci. Christian Faith, 44: 236-245.
Neel, J.V. (1982) "The Wonder of Our Presence Here: A Commentary on the Evolution and Maintenance of Human Diversity." Perspec. Biol. Med. 25: 518-558.
Rolston, H. (1992) "Smart Genes." Science and Religion News 3/1 Spring.
Romero-Herrera, A.E., Lieska, N., Goodman, M. and Simons, E.L. (1979) "The Use of Amino Acid Sequence Analysis in Assessing Evolution." Biochemie 61: 767-779.
Scherer, S. (1990) "The Protein Molecular Clock: Time for a Reevaluation." Evol. Biol. 24: 83-106.
Schwabe, C. (1986) "On the Validity of Molecular Evolution." Trends Biochem. Sci. 11: 280-286.
Shaw, J.P., Marks, J., Shen, C.C. and Shen C.K. (1989) "Anomolous and Selective DNA Mutations of the Old World Monkey." Proc. Nat. Acad. Sci. USA 86: 1312-1316.
Syvanen, M. (1986) "Cross-species Gene Transfer: A Major Factor in Evolution?" Trends. Genet. 2: 60-66.
Syvanen, M. (1987) "Molecular Clocks and Evolutionary Relationships: Possible Distortions Due to Horizontal Gene Flow." J. Mol. Evol. 26: 16-23.
Williams, J. (1974) "The Primary Structure of Proteins in Relation to Evolution." In: Chemistry of Macromolecules, H. Gutfreund (ed.), pp. 1-56. Baltimore, Md.: Univ. Park Press.
Yockey, H.P. (1977) "A Calculation of the Spontaneous Biogenesis by Information Theory." J. Theor. Biol. 67: 377-398.
Zuckerkandl, E. (1987) "On the Molecular Evolutionary Clock." J. Mol. Evol. 26: 34-46.
Zuckerkandl, E. and Pauling, L. (1962) "Molecular Disease, Evolution and Genetic Heterogeneity." In: Horizons in Biochemistry, M. Kasha and B. Pullman (Eds.), pp. 189-225. New York, N.Y.: Academic Press.